- immunodeficiency
- A condition resulting from a defective immune mechanism; may be primary (due to a defect in the immune mechanism itself) or secondary (dependent upon another disease process), specific (due to a defect in either the B-lymphocyte or the T-lymphocyte system, or both) or nonspecific (due to a defect in one or another component of the nonspecific immune mechanism : the complement, properdin, or phagocytic system). SYN: immune deficiency, immunity deficiency, immunologic deficiency.- cellular i. with abnormal immunoglobulin synthesis an ill-defined group of sporadic disorders of unknown cause, occurring in both males and females and associated with recurrent bacterial, fungal, protozoal, and viral infections; there is thymic hypoplasia with depressed cellular (T-lymphocyte) immunity combined with defective humoral (B-lymphocyte) immunity, although immunoglobulin levels may be normal. SYN: Nezelof syndrome.- combined i. i. of both the B-lymphocytes and T-lymphocytes.- common variable i. i. of unknown cause, and usually unclassifiable; usual onset after age 15 years but may occur at any age in either sex; the total quantity of immunoglobulin is commonly less than 300 mg/dL; the number of B lymphocytes is often within normal limits but there is a lack of plasma cells in lymphoid tissue; cellular (T-lymphocyte) immunity is usually intact; there is an increased susceptibility to pyogenic infection and often autoimmune disease. SYN: acquired agammaglobulinemia, acquired hypogammaglobulinemia.- phagocytic dysfunction i. suppression in number or function of phagocytic cells such as in chronic granulomatous disease. SYN: phagocytic dysfunction disorders i..- secondary i. i. in which there is no evident defect in the lymphoid tissues, but rather hypercatabolism or loss of immunoglobulins such as occurs in familial idiopathic hypercatabolic hypoproteinemia or in defects associated with the nephrotic syndrome. SYN: secondary agammaglobulinemia, secondary antibody deficiency, secondary hypogammaglobulinemia.- severe combined i. (SCID) [MIM*202500,MIM*300400, and MIM*312863] an i. in which there is absence of both humoral and cellular immunity with lymphopenia (of both B-type and T-type lymphocytes); characterized by thymus atrophy, lack of delayed hypersensitivity, and marked susceptibility to infections by bacteria, viruses, fungi, protozoa, and live vaccines; although bone marrow transplants have been effective, death may occur in the first year of life. Both autosomal recessive and X-linked forms occur; about one-half of those with autosomal recessive SCID have adenosine deaminase deficiency. The X-linked form is caused by mutation in the interleukin-2 receptor gamma gene (IL2RG) on Xq. SYN: Swiss type agammaglobulinemia.- i. with elevated IgM i. with reduced IgG- and IgA-bearing cells; there is recurrent pyogenic infection; X-linked in some families.
* * *
im·mu·no·de·fi·cien·cy -di-'fish-ən-sē n, pl -cies inability to produce a normal complement of antibodies or immunologically sensitized T cells esp. in response to specific antigens see AIDS* * *
n.deficiency in the immune response. This can be acquired, as in AIDS, but there are many varieties of primary immunodeficiency occurring as inherited disorders characterized by hypogammaglobulinaemia or defects in T-cell function, or both.* * *
im·mu·no·de·fi·cien·cy (im″u-no-də-fishґən-se) a deficiency of immune response or a disorder characterized by deficient immune response; classified as antibody (B cell), cellular (T cell), or combined immunodeficiency, or phagocytic dysfunction disorders. See accompanying table. See also acquired immunodeficiency syndrome, under syndrome. immunodeficient adj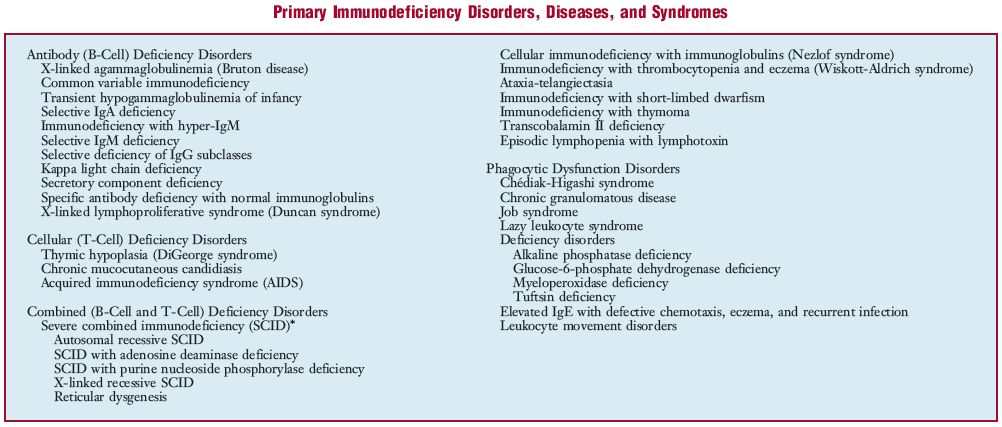
*Some forms of SCID, although deficient in both antibody and cellular immunity, actually have normal numbers of B cells.
Medical dictionary. 2011.