- Galactosemia
- A genetic metabolic disease in which there is a defect in the body's ability to use the sugar galactose. In classic galactosemia, the basic defect is a deficiency of the enzyme known as GALT (galactose-1-phosphate uridyl transferase). This causes an accumulation of galactose 1-phosphate which damages the liver, eye, brain and kidney. Galactosemia is one of the diseases in many newborn screening panels. The disease can be fatal, if undetected. If detected, it can be treated by avoiding galactose in the diet. Galactosemia is inherited as an autosomal recessive trait. There are two forms of the disease, GALT deficiency (classic galactosemia) and galactose kinase deficiency. Of the two, the GALT deficiency is the most severe. The GALT gene is in chromosome 9p13. People with galactosemia are unable to metabolize the simple sugar galactose. Galactose makes up half of the sugar called lactose that is found in milk. Lactose is called a disaccharide, di meaning 2, since lactose is made up of two sugars, galactose and glucose, bound together. If an infant with galactosemia is given milk, galactose builds up in the infants system causing damage to the liver, brain, kidneys and eyes. Individuals with galactosemis cannot tolerate any form of milk (human or otherwise) or any other galactose-containing food. Exposure to milk products will result in liver damage, mental retardation, cataract formation, and kidney failure. Typically, a newborn infant with galactosemia, upon being fed milk, will develop jaundice, vomiting, lethargy, irritability, and convulsions. The liver is enlarged and the blood sugar may be low. Continued feeding of milk products to the infant leads to cirrhosis of the liver, cataract formation in the eye resulting in partial blindness, and mental retardation. The symptoms of galactosemia include jaundice (yellowish discoloration of the skin and the whites of the eyes), vomiting, poor feeding, poor weight gain, lethargy, irritability, convulsions, and opacities in the lenses of the eyes. The signs detected include hepatomegaly (enlarged liver), hypoglycemia (low blood sugar), aminoaciduria (amino acids are present in the urine), cirrhosis, ascites (fluid collected within the abdomen), cataracts and mental retardation. The diagnosis is usually based on the demonstration of a lack of activity of the enzyme GALT in erythrocytes (red blood cells). Prenatal diagnosis is also feasible by direct measurement of the enzyme.
* * *1. [MIM*230400] An inborn error of galactose metabolism due to congenital deficiency of the enzyme galactosyl-1-phosphate uridylyltransferase, resulting in tissue accumulation of galactose 1-phosphate; manifested by nutritional failure, hepatosplenomegaly with cirrhosis, cataracts, mental retardation, galactosuria, aminoaciduria, and albuminuria that regress or disappear if galactose is removed from the diet; autosomal recessive inheritance; caused by mutation in the galactose-1-phosphate uridyltransferase gene (GALT) on 9p. SEE ALSO: galactokinase deficiency. 2. An inborn error in metabolism other than a deficiency in galactosyl-1-phosphate uridylyltransferase (see subentries below). SYN: galactose diabetes. [galactose + G. haima, blood]- epimerase deficiency g. an inborn error in metabolism in which there is a deficiency of uridine diphosphate galactose 4-epimerase; galactose 1-phosphate accumulates.- galactokinase deficiency g. an autosomal recessive disorder resulting in an accumulation of galactose and galactitol.- transferase deficiency g. an autosomal recessive disorder in which there is a deficiency of galactose-1-phosphate uridylyltransferase (see main entry for g.).
* * *
ga·lac·tos·emia or chiefly Brit ga·lac·tos·aemia gə-.lak-tə-'sē-mē-ə n a metabolic disorder inherited as an autosomal recessive trait in which galactose accumulates in the blood due to deficiency of an enzyme catalyzing its conversion to glucose* * *
ga·lac·tos·e·mia (gə-lak″to-seґme-ə) [galactose + -emia] a general term encompassing three autosomal recessive disorders resulting from defective galactose metabolism. Classic galactosemia, which is often fatal to neonates, is caused by mutations in the GALT gene (locus: 9p13), which encodes UDP-glucose–hexose-1-phosphate uridylyltransferase. Enzyme deficiency results in accumulation of galactose 1-phosphate and galactose, with cataracts, cirrhosis, hepatomegaly, vomiting, diarrhea, jaundice, poor weight gain, and malnutrition in infancy, and mental retardation in survivors. The other two disorders are galactokinase deficiency and UDP-glucose 4-epimerase deficiency (qq.v.).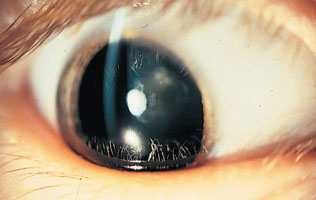
 Cataract associated with galactosemia, the opacity of the lens having an oil-droplet appearance.
Cataract associated with galactosemia, the opacity of the lens having an oil-droplet appearance.
Medical dictionary. 2011.